Researchers from Argonne National Laboratory, NVIDIA, the University of Chicago and more — awardees of a Gordon Bell special prize for COVID-19 research — developed a state-of-the-art model that processes genome-scale data.
by GEETIKA GUPTA
Editor’s note: This post was updated on November 17 after the announcement of the Gordon Bell prize winners.
The winner of the Gordon Bell special prize for high performance computing-based COVID-19 research has taught large language models (LLMs) a new lingo — gene sequences — that can unlock insights in genomics, epidemiology and protein engineering.
Published in October, the groundbreaking work is a collaboration by more than two dozen academic and commercial researchers from Argonne National Laboratory, NVIDIA, the University of Chicago and others.
The research team trained an LLM to track genetic mutations and predict variants of concern in SARS-CoV-2, the virus behind COVID-19. While most LLMs applied to biology to date have been trained on datasets of small molecules or proteins, this project is one of the first models trained on raw nucleotide sequences — the smallest units of DNA and RNA.
“We hypothesized that moving from protein-level to gene-level data might help us build better models to understand COVID variants,” said Arvind Ramanathan, computational biologist at Argonne, who led the project. “By training our model to track the entire genome and all the changes that appear in its evolution, we can make better predictions about not just COVID, but any disease with enough genomic data.”
The Gordon Bell awards, regarded as the Nobel Prize of high performance computing, were presented at the SC22 conference by the Association for Computing Machinery, which represents around 100,000 computing experts worldwide. Since 2020, the group has awarded a special prize for outstanding research that advances the understanding of COVID with HPC.
Training LLMs on a Four-Letter Language
LLMs have long been trained on human languages, which usually comprise a couple dozen letters that can be arranged into tens of thousands of words, and joined together into longer sentences and paragraphs. The language of biology, on the other hand, has only four letters representing nucleotides — A, T, G and C in DNA, or A, U, G and C in RNA — arranged into different sequences as genes.
While fewer letters may seem like a simpler challenge for AI, language models for biology are actually far more complicated. That’s because the genome — made up of over 3 billion nucleotides in humans, and about 30,000 nucleotides in coronaviruses — is difficult to break down into distinct, meaningful units.
“When it comes to understanding the code of life, a major challenge is that the sequencing information in the genome is quite vast,” Ramanathan said. “The meaning of a nucleotide sequence can be affected by another sequence that’s much further away than the next sentence or paragraph would be in human text. It could reach over the equivalent of chapters in a book.”
NVIDIA collaborators on the project designed a hierarchical diffusion method that enabled the LLM to treat long strings of around 1,500 nucleotides as if they were sentences.
“Standard language models have trouble generating coherent long sequences and learning the underlying distribution of different variants,” said paper co-author Anima Anandkumar, senior director of AI research at NVIDIA and Bren professor in the computing + mathematical sciences department at Caltech. “We developed a diffusion model that operates at a higher level of detail that allows us to generate realistic variants and capture better statistics.”
Predicting COVID Variants of Concern
Using open-source data from the Bacterial and Viral Bioinformatics Resource Center, the team first pretrained its LLM on more than 110 million gene sequences from prokaryotes, which are single-celled organisms like bacteria. It then fine-tuned the model using 1.5 million high-quality genome sequences for the COVID virus.
By pretraining on a broader dataset, the researchers also ensured their model could generalize to other prediction tasks in future projects — making it one of the first whole-genome-scale models with this capability.
Once fine-tuned on COVID data, the LLM was able to distinguish between genome sequences of the virus’ variants. It was also able to generate its own nucleotide sequences, predicting potential mutations of the COVID genome that could help scientists anticipate future variants of concern.

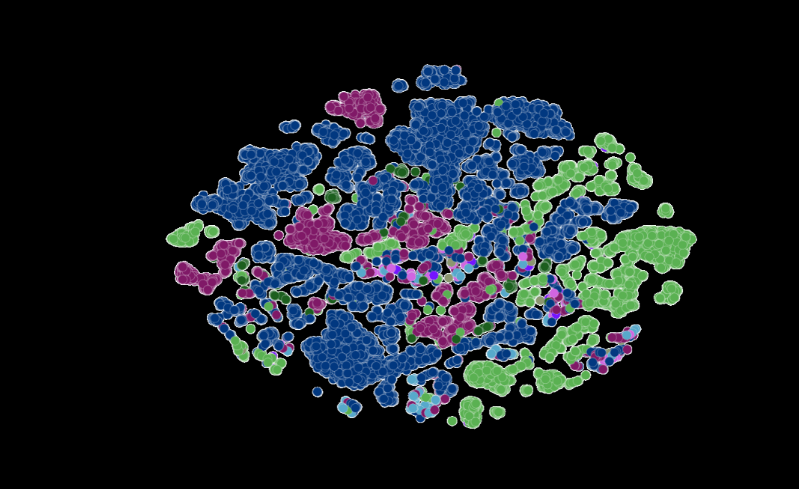
-Trained on a year’s worth of SARS-CoV-2 genome data, the model can infer the distinction between various viral strains. Each dot on the left corresponds to a sequenced SARS-CoV-2 viral strain, color-coded by variant.
The figure on the right zooms into one particular strain of the virus, which captures evolutionary couplings across the viral proteins specific to this strain. Image courtesy of Argonne National Laboratory’s Bharat Kale, Max Zvyagin and Michael E. Papka.
“Most researchers have been tracking mutations in the spike protein of the COVID virus, specifically the domain that binds with human cells,” Ramanathan said. “But there are other proteins in the viral genome that go through frequent mutations and are important to understand.”
The model could also integrate with popular protein-structure-prediction models like AlphaFold and OpenFold, the paper stated, helping researchers simulate viral structure and study how genetic mutations impact a virus’ ability to infect its host. OpenFold is one of the pretrained language models included in the NVIDIA BioNeMo LLM service for developers applying LLMs to digital biology and chemistry applications.
Supercharging AI Training With GPU-Accelerated Supercomputers
The team developed its AI models on supercomputers powered by NVIDIA A100 Tensor Core GPUs — including Argonne’s Polaris, the U.S. Department of Energy’s Perlmutter, and NVIDIA’s in-house Selene system. By scaling up to these powerful systems, they achieved performance of more than 1,500 exaflops in training runs, creating the largest biological language models to date.
“We’re working with models today that have up to 25 billion parameters, and we expect this to significantly increase in the future,” said Ramanathan. “The model size, the genetic sequence lengths and the amount of training data needed means we really need the computational complexity provided by supercomputers with thousands of GPUs.”
The researchers estimate that training a version of their model with 2.5 billion parameters took over a month on around 4,000 GPUs. The team, which was already investigating LLMs for biology, spent about four months on the project before publicly releasing the paper and code. The GitHub page includes instructions for other researchers to run the model on Polaris and Perlmutter.
The NVIDIA BioNeMo framework, available in early access on the NVIDIA NGC hub for GPU-optimized software, supports researchers scaling large biomolecular language models across multiple GPUs. Part of the NVIDIA Clara Discovery collection of drug discovery tools, the framework will support chemistry, protein, DNA and RNA data formats.
Find NVIDIA at SC22 and watch a replay of the special address below:
Image at top represents COVID strains sequenced by the researchers’ LLM. Each dot is color-coded by COVID variant. Image courtesy of Argonne National Laboratory’s Bharat Kale, Max Zvyagin and Michael E. Papka.
Researchers from Argonne National Laboratory, NVIDIA, the University of Chicago and more — awardees of a Gordon Bell special prize for COVID-19 research — developed a state-of-the-art model that processes genome-scale data.
by GEETIKA GUPTA
Editor’s note: This post was updated on November 17 after the announcement of the Gordon Bell prize winners.
The winner of the Gordon Bell special prize for high performance computing-based COVID-19 research has taught large language models (LLMs) a new lingo — gene sequences — that can unlock insights in genomics, epidemiology and protein engineering.
Published in October, the groundbreaking work is a collaboration by more than two dozen academic and commercial researchers from Argonne National Laboratory, NVIDIA, the University of Chicago and others.
The research team trained an LLM to track genetic mutations and predict variants of concern in SARS-CoV-2, the virus behind COVID-19. While most LLMs applied to biology to date have been trained on datasets of small molecules or proteins, this project is one of the first models trained on raw nucleotide sequences — the smallest units of DNA and RNA.
“We hypothesized that moving from protein-level to gene-level data might help us build better models to understand COVID variants,” said Arvind Ramanathan, computational biologist at Argonne, who led the project. “By training our model to track the entire genome and all the changes that appear in its evolution, we can make better predictions about not just COVID, but any disease with enough genomic data.”
The Gordon Bell awards, regarded as the Nobel Prize of high performance computing, were presented at the SC22 conference by the Association for Computing Machinery, which represents around 100,000 computing experts worldwide. Since 2020, the group has awarded a special prize for outstanding research that advances the understanding of COVID with HPC.
Training LLMs on a Four-Letter Language
LLMs have long been trained on human languages, which usually comprise a couple dozen letters that can be arranged into tens of thousands of words, and joined together into longer sentences and paragraphs. The language of biology, on the other hand, has only four letters representing nucleotides — A, T, G and C in DNA, or A, U, G and C in RNA — arranged into different sequences as genes.
While fewer letters may seem like a simpler challenge for AI, language models for biology are actually far more complicated. That’s because the genome — made up of over 3 billion nucleotides in humans, and about 30,000 nucleotides in coronaviruses — is difficult to break down into distinct, meaningful units.
“When it comes to understanding the code of life, a major challenge is that the sequencing information in the genome is quite vast,” Ramanathan said. “The meaning of a nucleotide sequence can be affected by another sequence that’s much further away than the next sentence or paragraph would be in human text. It could reach over the equivalent of chapters in a book.”
NVIDIA collaborators on the project designed a hierarchical diffusion method that enabled the LLM to treat long strings of around 1,500 nucleotides as if they were sentences.
“Standard language models have trouble generating coherent long sequences and learning the underlying distribution of different variants,” said paper co-author Anima Anandkumar, senior director of AI research at NVIDIA and Bren professor in the computing + mathematical sciences department at Caltech. “We developed a diffusion model that operates at a higher level of detail that allows us to generate realistic variants and capture better statistics.”
Predicting COVID Variants of Concern
Using open-source data from the Bacterial and Viral Bioinformatics Resource Center, the team first pretrained its LLM on more than 110 million gene sequences from prokaryotes, which are single-celled organisms like bacteria. It then fine-tuned the model using 1.5 million high-quality genome sequences for the COVID virus.
By pretraining on a broader dataset, the researchers also ensured their model could generalize to other prediction tasks in future projects — making it one of the first whole-genome-scale models with this capability.
Once fine-tuned on COVID data, the LLM was able to distinguish between genome sequences of the virus’ variants. It was also able to generate its own nucleotide sequences, predicting potential mutations of the COVID genome that could help scientists anticipate future variants of concern.
-Trained on a year’s worth of SARS-CoV-2 genome data, the model can infer the distinction between various viral strains. Each dot on the left corresponds to a sequenced SARS-CoV-2 viral strain, color-coded by variant.
The figure on the right zooms into one particular strain of the virus, which captures evolutionary couplings across the viral proteins specific to this strain. Image courtesy of Argonne National Laboratory’s Bharat Kale, Max Zvyagin and Michael E. Papka.
“Most researchers have been tracking mutations in the spike protein of the COVID virus, specifically the domain that binds with human cells,” Ramanathan said. “But there are other proteins in the viral genome that go through frequent mutations and are important to understand.”
The model could also integrate with popular protein-structure-prediction models like AlphaFold and OpenFold, the paper stated, helping researchers simulate viral structure and study how genetic mutations impact a virus’ ability to infect its host. OpenFold is one of the pretrained language models included in the NVIDIA BioNeMo LLM service for developers applying LLMs to digital biology and chemistry applications.
Supercharging AI Training With GPU-Accelerated Supercomputers
The team developed its AI models on supercomputers powered by NVIDIA A100 Tensor Core GPUs — including Argonne’s Polaris, the U.S. Department of Energy’s Perlmutter, and NVIDIA’s in-house Selene system. By scaling up to these powerful systems, they achieved performance of more than 1,500 exaflops in training runs, creating the largest biological language models to date.
“We’re working with models today that have up to 25 billion parameters, and we expect this to significantly increase in the future,” said Ramanathan. “The model size, the genetic sequence lengths and the amount of training data needed means we really need the computational complexity provided by supercomputers with thousands of GPUs.”
The researchers estimate that training a version of their model with 2.5 billion parameters took over a month on around 4,000 GPUs. The team, which was already investigating LLMs for biology, spent about four months on the project before publicly releasing the paper and code. The GitHub page includes instructions for other researchers to run the model on Polaris and Perlmutter.
The NVIDIA BioNeMo framework, available in early access on the NVIDIA NGC hub for GPU-optimized software, supports researchers scaling large biomolecular language models across multiple GPUs. Part of the NVIDIA Clara Discovery collection of drug discovery tools, the framework will support chemistry, protein, DNA and RNA data formats.
Find NVIDIA at SC22 and watch a replay of the special address below:
Image at top represents COVID strains sequenced by the researchers’ LLM. Each dot is color-coded by COVID variant. Image courtesy of Argonne National Laboratory’s Bharat Kale, Max Zvyagin and Michael E. Papka.